The scSpecies workflow
This tutorial demonstrates how to apply the scSpecies workflow to align three scRNA-seq datasets (mice, humans and hamsters).
We start by loading the preprocesed .h5mu file saved within the data_prepocessing.ipynb notebook.
[1]:
import os
import muon as mu
mu.set_options(pull_on_update=False)
path = os.path.abspath('').replace('\\', '/')+'/'
mdata = mu.read_h5mu(path+'data/liver_atlas.h5mu')
# For coloring
mdata.mod['human'].obs['cell_type_fine'] = mdata.mod['human'].obs['cell_type_fine'].cat.rename_categories({'Naive-CM CD4+ T': 'Naive/CM CD4+ T'})
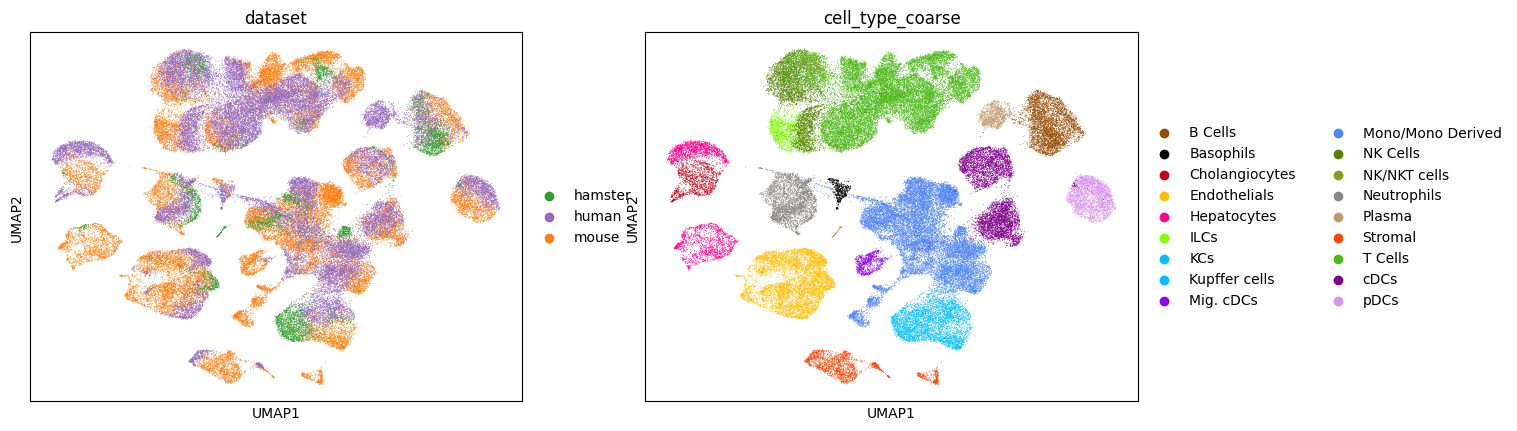
Before we pre-train scSpecies, we plot a UMAP representation of the unaligned mouse/human dataset pair on the data-level.
[2]:
from scipy import sparse
import anndata as ad
import scanpy as sc
import pandas as pd
import numpy as np
from scspecies.plot import return_palette
from scspecies.models import neighbors_workaround
# Subsetting to homologous genes
_, hom_ind_mouse, hom_ind_human = np.intersect1d(mdata.mod['mouse'].var_names, mdata.mod['human'].var['var_names_transl'], return_indices=True)
adata_concat = ad.AnnData(
X=sparse.vstack([mdata.mod['mouse'][:, hom_ind_mouse].X, mdata.mod['human'][:, hom_ind_human].X]).toarray(),
obs=pd.concat([mdata.mod['mouse'].obs, mdata.mod['human'].obs])
).copy()
# Color scheme for the liver cell dataset. Won't return nice results for other datasets.
palette = return_palette(list(adata_concat.obs.cell_type_fine.unique()) + list(adata_concat.obs.dataset.unique()))
sc.pp.pca(adata_concat)
sc.pp.neighbors(adata_concat, use_rep='X_pca')
# sc.pp.neighbors can crash the kernel for M1/M2 chips. In this case use a workaroung for this function.
#neighbors_workaround(adata_concat, use_rep='X_pca')
sc.tl.umap(adata_concat)
sc.pl.umap(adata_concat, color=['dataset', 'cell_type_fine'], palette=palette)
1) Context and target dataset alignment
scSpecies class.[3]:
from scspecies.models import scSpecies
import torch
device = ("mps" if torch.backends.mps.is_available() and torch.backends.mps.is_built() else "cuda" if torch.cuda.is_available() else "cpu")
model = scSpecies(device,
mdata,
path,
context_key = 'mouse',
target_key = 'human',
random_seed=1234
)
Initializing context scVI model.
Initializing target scVI model.
train_context for 30 epochs and save the model parameters to path.get_representation an save them in the context modality in the .obsm layer.[4]:
model.train_context(30, save_key='_mouse')
model.get_representation(eval_model='context', save_intermediate=True, save_libsize=True)
Pretraining on the context dataset for 30 epochs (= 8820 iterations).
Progress: 99.8% - ETA: 0:00:00 - Epoch: 30 - Iteration: 8800 - ms/Iteration: 15.80 - nELBO: 1461.4 (+1.410) - nlog_likeli: 1443.2 (+1.366) - KL-Div z: 15.140 (+0.023) - KL-Div l: 3.0712 (+0.022). Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/config_dict.pkl
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_config__mouse.pkl
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_optimizer__mouse.opt
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_encoder_inner__mouse.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_encoder_outer__mouse.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_decoder__mouse.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_lib_encoder__mouse.pth.
Calculate latent variables. Step 242/295
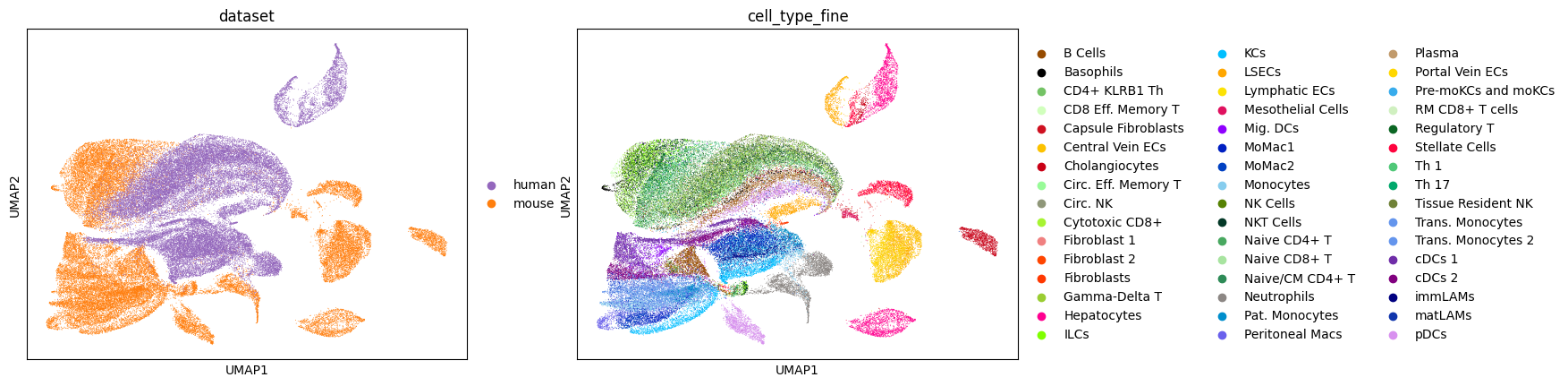
We check that the context scVI model has separated latent cell clusters by visualizing the latent representation with UMAP.
[5]:
sc.pp.neighbors(model.mdata['mouse'], use_rep='z_mu')
#neighbors_workaround(model.mdata['mouse'], use_rep='z_mu')
sc.tl.umap(model.mdata['mouse'])
sc.pl.umap(model.mdata['mouse'], color='cell_type_fine', palette=palette)
Next we fine-tune by training the target scVI model and aligning the human dataset with this latent representations.
[6]:
model.train_target(30, save_key='_human')
model.get_representation(eval_model='target', save_libsize=True)
Training on the target dataset for 30 epochs (= 8190 iterations).
Progress: 100.0% - ETA: 0:00:00 - Epoch: 30 - Iteration: 8187 - ms/Iteration: 36.19 - nELBO: 1847.1 (+0.706) - nlog_likeli: 1384.8 (+0.496) - KL-Div z: 13.803 (+0.006) - KL-Div l: 2.8112 (+0.021) - Align-Term: 445.69 (+0.183). Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/config_dict.pkl
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_config__human.pkl
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_optimizer__human.opt
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_encoder_inner__human.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_encoder_outer__human.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_decoder__human.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_lib_encoder__human.pth.
Calculate latent variables. Step 181/274
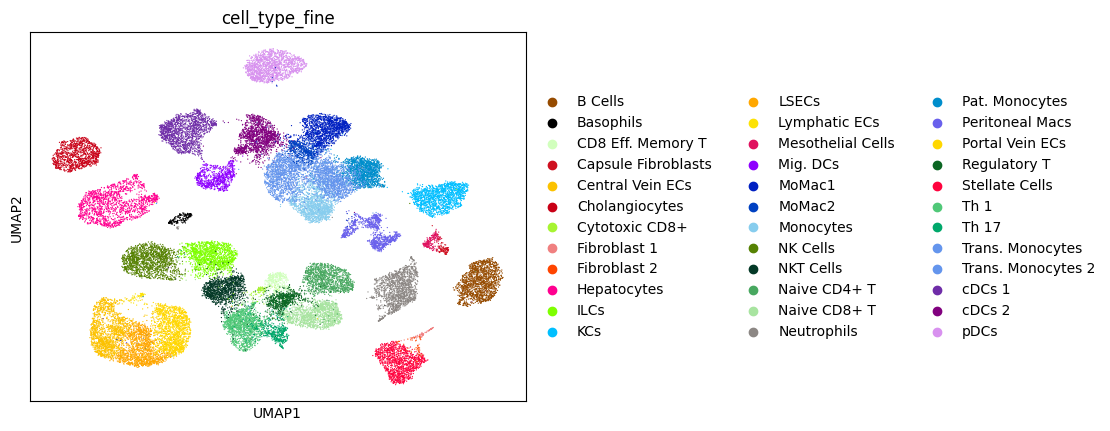
After fine-tuning, we can visualize the aligned latent representation.
[7]:
adata_concat.obsm['lat_rep'] = np.concat([mdata.mod['mouse'].obsm['z_mu'], mdata.mod['human'].obsm['z_mu']])
sc.pp.neighbors(adata_concat, use_rep='lat_rep')
#neighbors_workaround(adata_concat, use_rep='lat_rep')
sc.tl.umap(adata_concat)
sc.pl.umap(adata_concat, color=['dataset', 'cell_type_fine'], palette=palette)
2) Information transfer via similarity scores
We continue by evaluating the likelihood based similarity measure between target and context cell types with return_similarity_df_prot.
scale=min_max or scale=max the scores are scaled such that the values can be interpreted as 1 = most similar. Without scaling the highest values (closest to zero) represent high similarity. The output dataframe contains target cell types in df.index and context cell types in df.columns.We can use these similarity scores to match cell type annotation between datasets. To reduce computational demand reduce the sample size via max_sample_targ and max_sample_cont.
[8]:
from scspecies.plot import plot_prototype_sim_heatmap
import matplotlib.pyplot as plt
import seaborn as sns
df = model.return_similarity_df()
print(df.head())
plot_prototype_sim_heatmap(df)
1152/1152 Similarity calculation for the pDCs-pDCs pair B Cells Basophils CD8 Eff. Memory T \
B Cells -3.551205 -347.071991 -205.805511
Basophils -367.502869 -6.766159 -367.215576
CD4+ KLRB1 Th -215.161743 -271.281860 -78.929642
Central Vein ECs -322.188660 -700.436768 -326.259460
Cholangiocytes -499.112000 -485.331177 -334.543152
Capsule Fibroblasts Central Vein ECs Cholangiocytes \
B Cells -194.542191 -367.720825 -181.570511
Basophils -294.482727 -346.947021 -234.808044
CD4+ KLRB1 Th -171.188400 -399.845886 -157.499298
Central Vein ECs -422.906860 -49.124344 -588.992798
Cholangiocytes -310.032990 -401.235352 -18.102785
Cytotoxic CD8+ Fibroblast 1 Fibroblast 2 Hepatocytes \
B Cells -276.343140 -300.546570 -321.969421 -171.634369
Basophils -354.181030 -196.589905 -237.053650 -226.197464
CD4+ KLRB1 Th -43.449066 -243.138275 -263.490784 -173.533569
Central Vein ECs -316.847839 -387.683533 -250.165573 -651.404297
Cholangiocytes -311.309723 -368.605804 -202.102081 -515.239136
... Portal Vein ECs Regulatory T Stellate Cells \
B Cells ... -287.618103 -188.015732 -199.216400
Basophils ... -294.696503 -322.972168 -277.173767
CD4+ KLRB1 Th ... -109.869568 -46.969368 -84.444969
Central Vein ECs ... -87.446487 -325.616760 -229.602570
Cholangiocytes ... -408.232971 -329.584839 -336.466187
Th 1 Th 17 Trans. Monocytes \
B Cells -477.406464 -164.725906 -254.275375
Basophils -345.458740 -289.259583 -350.966553
CD4+ KLRB1 Th -19.942635 -21.023905 -369.132538
Central Vein ECs -258.104187 -218.185822 -927.046509
Cholangiocytes -354.853943 -267.215668 -1089.590820
Trans. Monocytes 2 cDCs 1 cDCs 2 pDCs
B Cells -277.881897 -266.351868 -209.525513 -215.003510
Basophils -380.898071 -426.963593 -342.693695 -366.804199
CD4+ KLRB1 Th -380.841309 -364.149567 -327.140259 -357.559448
Central Vein ECs -953.276672 -519.542603 -533.386597 -706.463196
Cholangiocytes -897.322388 -430.416077 -662.558472 -510.795807
[5 rows x 36 columns]
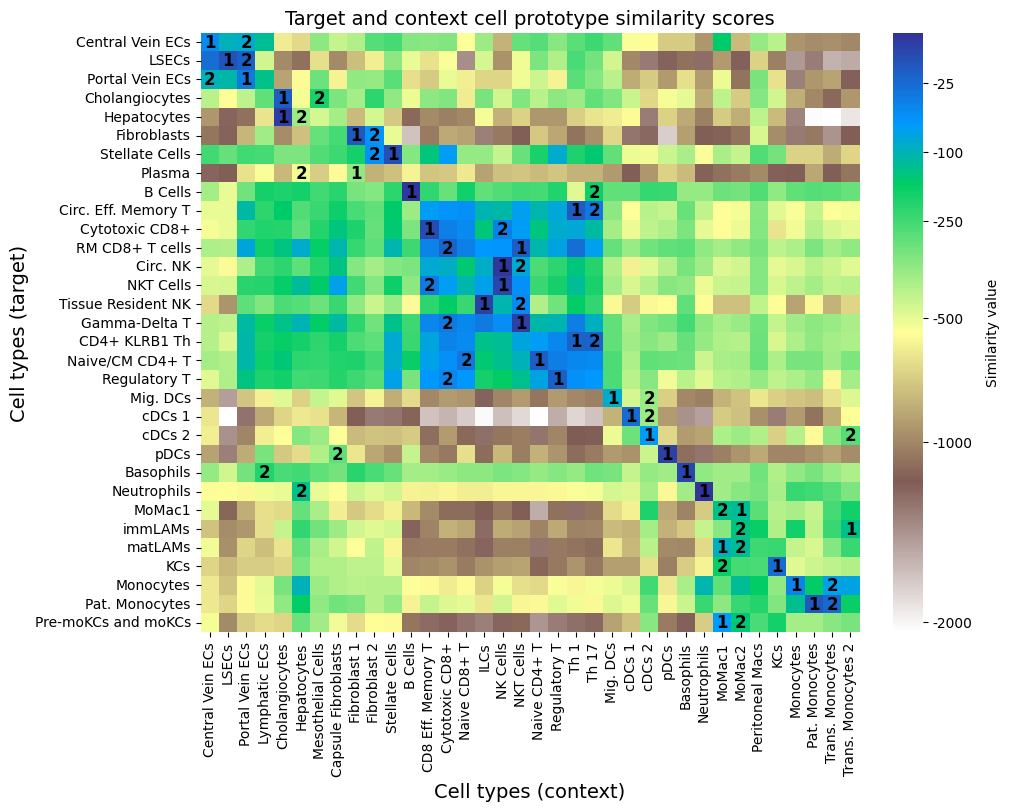
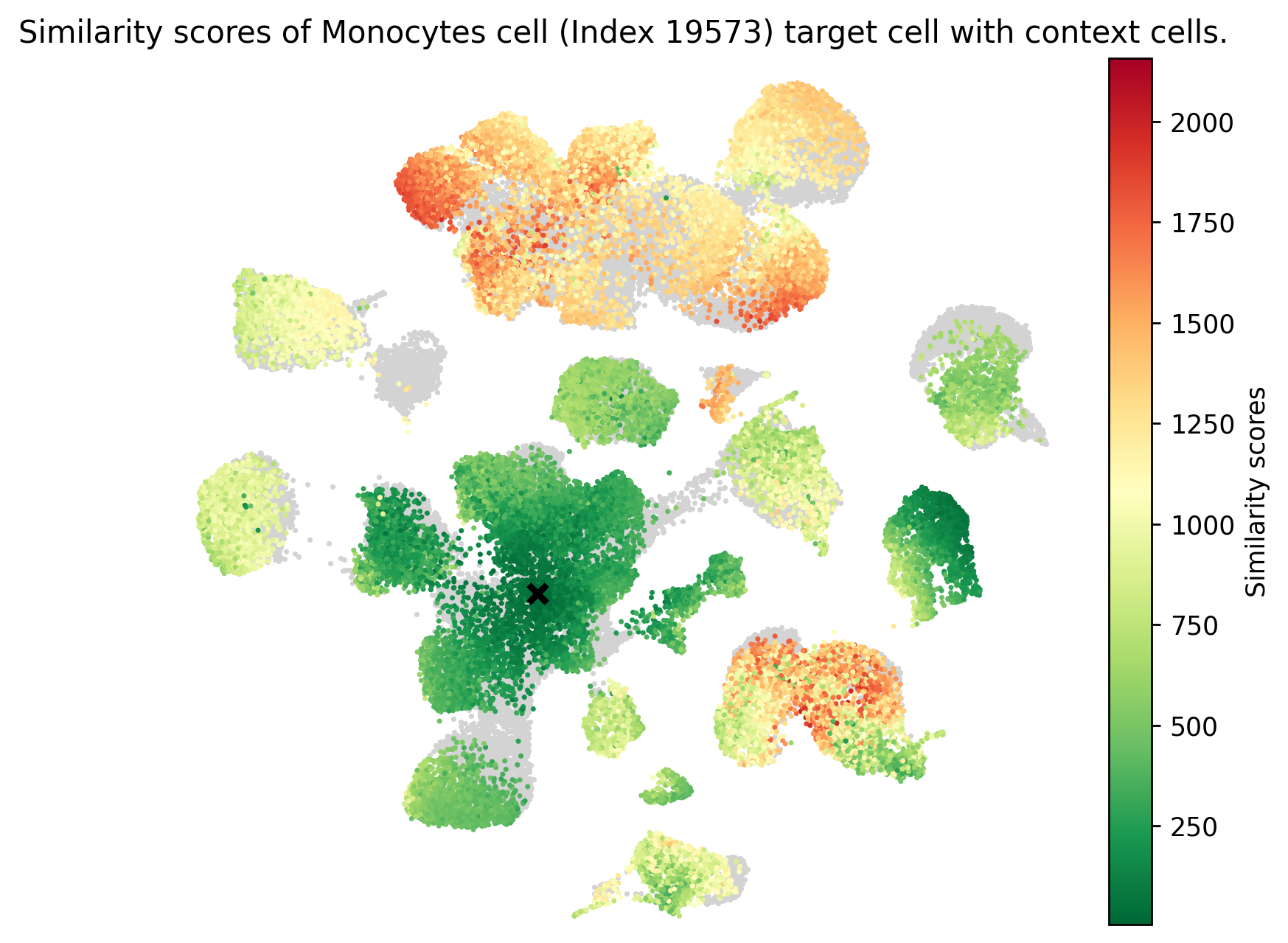
We can use the similarity measure to infer target label annotation from context cells.
transfer_labels_cell..obs labels specified in context_obs_transfer.df_neigbor dataframe contains all context cells with corresponding indices sorted by similarity.plot_similarity.[9]:
from scspecies.plot import plot_similarity
human_cell_types = model.mdata['human'].obs['cell_type_fine']
mouse_cell_types = model.mdata['mouse'].obs['cell_type_fine']
common_cell_types = np.intersect1d(human_cell_types.unique(), mouse_cell_types.unique())
human_inds = human_cell_types.isin(common_cell_types).to_numpy().nonzero()[0]
human_ind = np.random.choice(human_inds)
context_obs_transfer = ['cell_type_coarse', 'cell_type_fine']
df_neigbor = model.transfer_labels_cell(human_ind, context_obs_transfer)
print('Index of target human cell: {}, Information: {}.'.format(str(human_ind), ', '.join([obs_name+': '+label for label, obs_name in zip(model.mdata['human'].obs[context_obs_transfer].iloc[human_ind].values[0], context_obs_transfer)])))
print(df_neigbor.head())
plot_similarity(adata_concat, df_neigbor, human_ind)
Index of target human cell: 19573, Information: cell_type_coarse: M, cell_type_fine: o.
cell_type_coarse cell_type_fine index \
GCAGCCATCGTGGGAA-46_mouse Mono-Mono Derived Trans. Monocytes 2 30597
CGGACTGGTGACTCAT-42_mouse Mono-Mono Derived Trans. Monocytes 2 30881
TCAATCTCATTTGCCC-41_mouse Mono-Mono Derived Trans. Monocytes 2 31607
CCTCTGATCGGTTCGG-46_mouse Mono-Mono Derived Trans. Monocytes 2 31386
CTCTAATCACCATGTA-41_mouse Mono-Mono Derived Trans. Monocytes 2 31559
similarity_score
GCAGCCATCGTGGGAA-46_mouse 5.018066
CGGACTGGTGACTCAT-42_mouse 7.247314
TCAATCTCATTTGCCC-41_mouse 7.400024
CCTCTGATCGGTTCGG-46_mouse 7.458252
CTCTAATCACCATGTA-41_mouse 8.483887
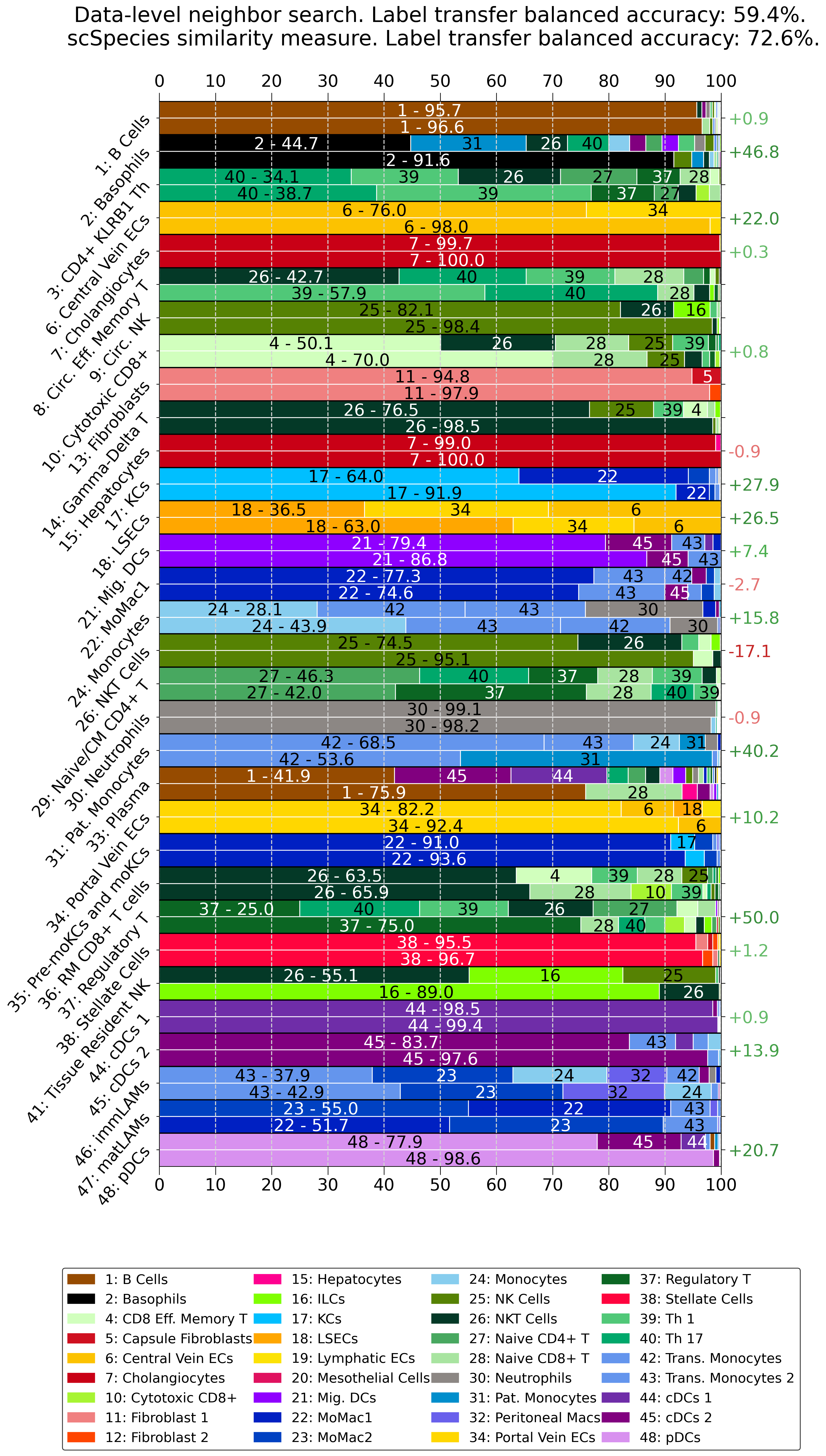
transfer_labels_dataset..obs layer of the target dataset.ret_pred_df.[10]:
context_obs_transfer = ['cell_type_coarse', 'cell_type_fine']
model.transfer_labels_data(context_obs_transfer)
print(model.mdata['human'].obs[['pred_sim_cell_type_coarse', 'cell_type_coarse', 'pred_sim_cell_type_fine', 'cell_type_fine']].head())
df_nns, bas_nns = model.ret_pred_df(pred_key='pred_nns_cell_type_fine', target_label_key='cell_type_fine', context_label_key='cell_type_fine')
df_sim, bas_sim = model.ret_pred_df(pred_key='pred_sim_cell_type_fine', target_label_key='cell_type_fine', context_label_key='cell_type_fine')
print(df_sim.head())
print('Data-level k=25 nearest neighbor search --> Balanced accuracy: {}%'.format(round(bas_nns*100,2)))
print('Label tarnsfer using similarity measure --> Balanced accuracy: {}%'.format(round(bas_sim*100,2)))
Pre-computing latent space NNS with 250 neighbors using the euclidean distance.
Calculate similarity metric. Step 273/274. pred_sim_cell_type_coarse cell_type_coarse \
TTCGGTCCACGGCCAT-12_human Mono-Mono Derived Mono-Mono Derived
TCATTTGCAGTCAGAG-1_human Mono-Mono Derived Mono-Mono Derived
TAGTTGGCATCCCACT-4_human Mono-Mono Derived Mono-Mono Derived
GGATTACTCCTCGCAT-12_human Mono-Mono Derived Mono-Mono Derived
TGGTAGTGTGGCTACC-28_human Mono-Mono Derived Mono-Mono Derived
pred_sim_cell_type_fine cell_type_fine
TTCGGTCCACGGCCAT-12_human MoMac1 Pre-moKCs and moKCs
TCATTTGCAGTCAGAG-1_human MoMac1 Pre-moKCs and moKCs
TAGTTGGCATCCCACT-4_human MoMac1 Pre-moKCs and moKCs
GGATTACTCCTCGCAT-12_human MoMac1 Pre-moKCs and moKCs
TGGTAGTGTGGCTACC-28_human MoMac1 Pre-moKCs and moKCs
/Users/cschaech/anaconda3/envs/scspecies-env2/lib/python3.11/site-packages/sklearn/metrics/_classification.py:2801: UserWarning: y_pred contains classes not in y_true
warnings.warn("y_pred contains classes not in y_true")
/Users/cschaech/anaconda3/envs/scspecies-env2/lib/python3.11/site-packages/sklearn/metrics/_classification.py:2801: UserWarning: y_pred contains classes not in y_true
warnings.warn("y_pred contains classes not in y_true")
B Cells Basophils CD8 Eff. Memory T Capsule Fibroblasts \
B Cells 96.6 0.000000 0.000000 0.0
Basophils 0.0 91.578947 0.000000 0.0
CD4+ KLRB1 Th 0.0 0.000000 0.066667 0.0
Central Vein ECs 0.0 0.000000 0.000000 0.0
Cholangiocytes 0.0 0.000000 0.000000 0.0
Central Vein ECs Cholangiocytes Cytotoxic CD8+ \
B Cells 0.0 0.0 0.000000
Basophils 0.0 0.0 0.000000
CD4+ KLRB1 Th 0.0 0.0 2.333333
Central Vein ECs 98.0 0.0 0.000000
Cholangiocytes 0.0 100.0 0.000000
Fibroblast 1 Fibroblast 2 Hepatocytes ... \
B Cells 0.0 0.0 0.0 ...
Basophils 0.0 0.0 0.0 ...
CD4+ KLRB1 Th 0.0 0.0 0.0 ...
Central Vein ECs 0.0 0.0 0.0 ...
Cholangiocytes 0.0 0.0 0.0 ...
Portal Vein ECs Regulatory T Stellate Cells Th 1 \
B Cells 0.133333 0.066667 0.000000 0.0
Basophils 0.000000 0.000000 0.000000 0.0
CD4+ KLRB1 Th 0.000000 11.200000 0.066667 38.2
Central Vein ECs 2.000000 0.000000 0.000000 0.0
Cholangiocytes 0.000000 0.000000 0.000000 0.0
Th 17 Trans. Monocytes Trans. Monocytes 2 cDCs 1 \
B Cells 0.000000 0.4 0.0 0.0
Basophils 0.263158 0.0 0.0 0.0
CD4+ KLRB1 Th 38.666667 0.0 0.0 0.0
Central Vein ECs 0.000000 0.0 0.0 0.0
Cholangiocytes 0.000000 0.0 0.0 0.0
cDCs 2 pDCs
B Cells 0.200000 0.0
Basophils 0.263158 0.0
CD4+ KLRB1 Th 0.000000 0.0
Central Vein ECs 0.000000 0.0
Cholangiocytes 0.000000 0.0
[5 rows x 36 columns]
Data-level k=25 nearest neighbor search --> Balanced accuracy: 59.36%
Label tarnsfer using similarity measure --> Balanced accuracy: 72.56%
[11]:
from scspecies.plot import label_transfer_acc
label_transfer_acc(df_nns, df_sim)
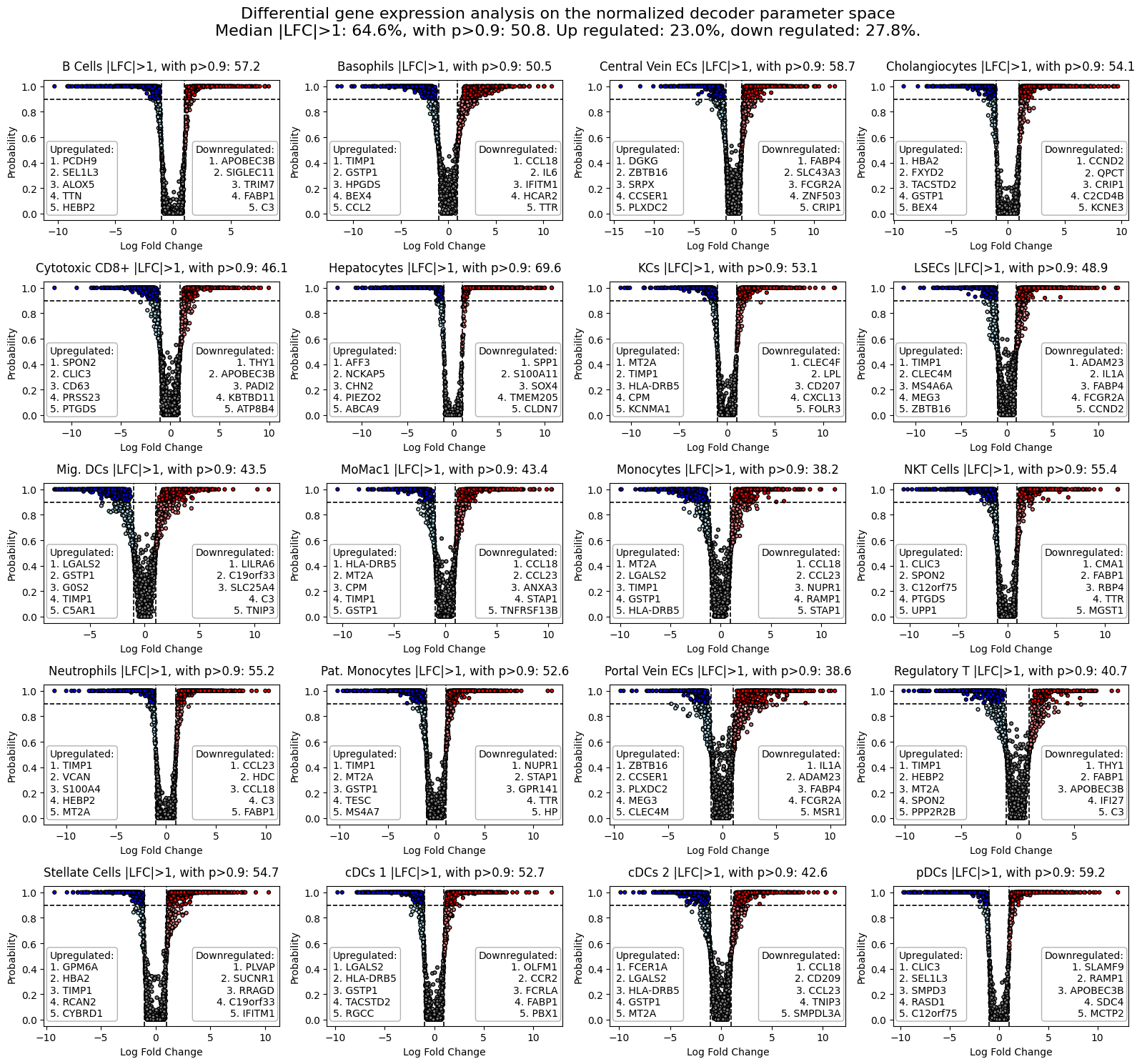
3) Differential gene expression analysis
The difference in modeled gene expression can be analyzed by comparing the log2-fold change in normalized gene expression with compute_logfold_change.
[12]:
lfc_dict = model.compute_logfold_change(lfc_delta = 1)
The output is a dictionary with cell type wise data frames containing logfoldchange values and other information. The dataframes contain the homologous traget gene symbols in their index and their chosen labels as columns.
rho_median_context: Contains median context normalized gene expression,mu_median_context: Contains median context expected value gene expression,rho_median_target: Contains median target normalized gene expression,mu_median_target: Contains median target expected value gene expression,lfc: Contains mMedian Log2 fold-change of the relative expression parameter rho,p: Probability of Log2 fold-change values greater thanlfc_delta,lfc_rand: Contains median Log2 fold-change of the relative expression parameter rho on permuted data,p_rand: Probability of Log2 fold-change values greater thanlfc_deltaon permuted data.
[13]:
print('Results for', list(lfc_dict.keys())[0])
lfc_dict[list(lfc_dict.keys())[0]].head()
Results for B Cells
[13]:
| rho_median_context | mu_median_context | rho_median_target | mu_median_target | lfc | p | lfc_rand | p_rand | |
|---|---|---|---|---|---|---|---|---|
| C12orf75 | 1.206251e-07 | 0.000181 | 0.000089 | 0.123426 | 6.322998 | 1.000000 | -5.483539 | 1.000000 |
| C1orf21 | 6.015457e-06 | 0.009070 | 0.000006 | 0.007996 | -0.119253 | 0.021987 | -4.536520 | 1.000000 |
| C19orf33 | 2.811825e-05 | 0.041576 | 0.000001 | 0.001560 | -3.784217 | 1.000000 | -5.864892 | 1.000000 |
| KIAA0513 | 1.824987e-05 | 0.028230 | 0.000008 | 0.012216 | -1.027371 | 0.539969 | 0.696235 | 0.173268 |
| C19orf38 | 5.501264e-04 | 0.839834 | 0.000026 | 0.036723 | -4.331485 | 1.000000 | -4.134716 | 1.000000 |
We can visualize the results with plot_lfc per cell type.
[14]:
from scspecies.plot import plot_lfc
plot_lfc(lfc_dict)
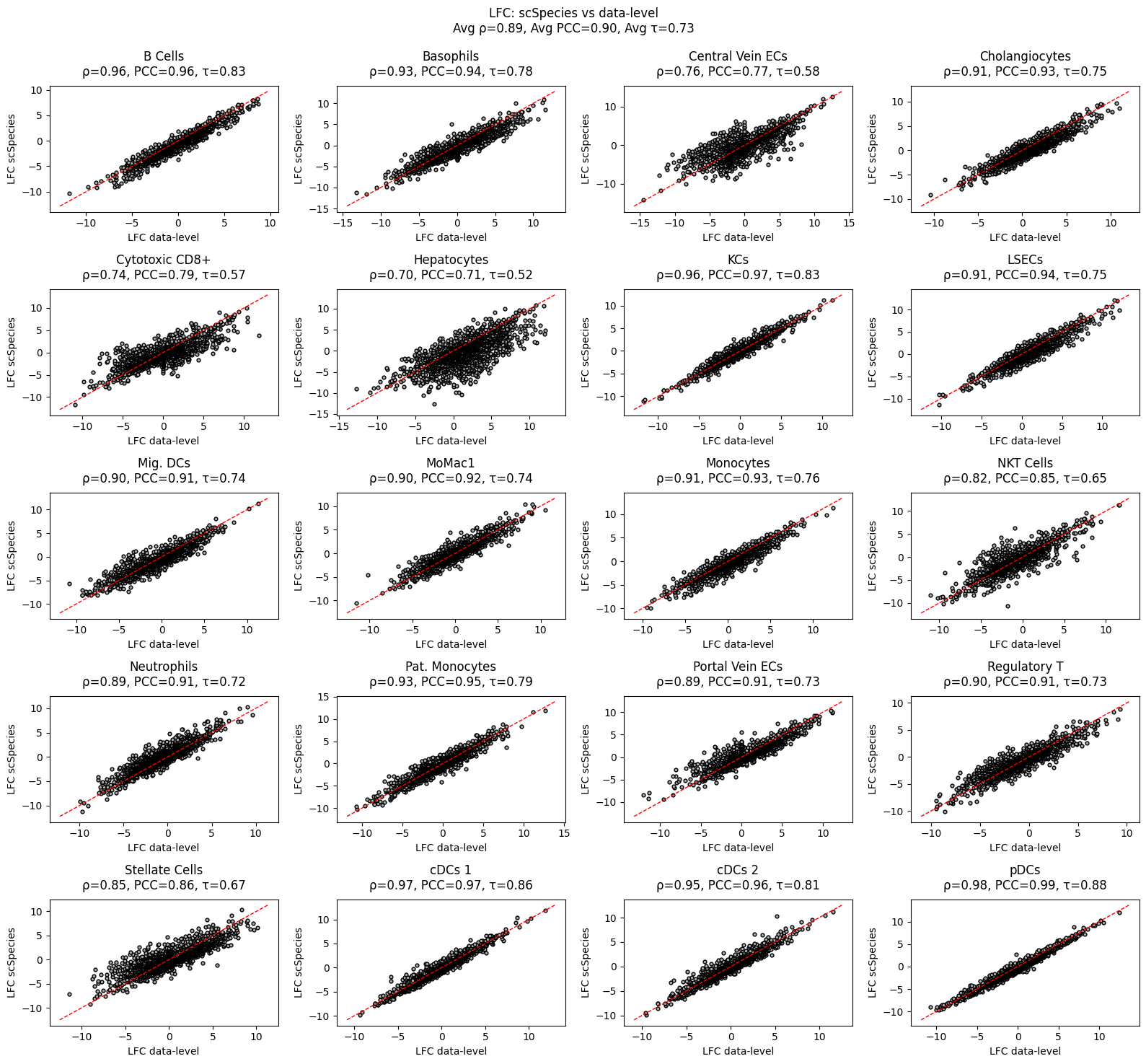
Lets compare the results with a DFG Analysis at the data level. For this we generate homologous cell samples from the latent space.
[15]:
from scspecies.plot import plot_lfc_comparison
target_rho_dict, context_rho_dict = model.generate_homologous_samples(samples=2000)
plot_lfc_comparison(model, lfc_dict)
4) Creating a cell atlas
NOTE: For differential gene expression analysis the context decoder should be retrained.
[17]:
# Just for coloring
mdata.mod['human'].obs['cell_type_coarse'] = mdata.mod['human'].obs['cell_type_coarse'].cat.rename_categories({'Mono-Mono Derived': 'Mono/Mono Derived'})
mdata.mod['mouse'].obs['cell_type_coarse'] = mdata.mod['mouse'].obs['cell_type_coarse'].cat.rename_categories({'Mono-Mono Derived': 'Mono/Mono Derived'})
mdata.mod['hamster'].obs['cell_type_coarse'] = mdata.mod['hamster'].obs['cell_type_coarse'].cat.rename_categories({'Mono-Mono Derived': 'Mono/Mono Derived'})
mdata.mod['human'].obs['cell_type_coarse'] = mdata.mod['human'].obs['cell_type_coarse'].cat.rename_categories({'NK-NKT cells': 'NK/NKT cells'})
mdata.mod['mouse'].obs['cell_type_coarse'] = mdata.mod['mouse'].obs['cell_type_coarse'].cat.rename_categories({'NK-NKT cells': 'NK/NKT cells'})
mdata.mod['hamster'].obs['cell_type_coarse'] = mdata.mod['hamster'].obs['cell_type_coarse'].cat.rename_categories({'NK-NKT cells': 'NK/NKT cells'})
model_hamster = scSpecies(device,
mdata,
path,
context_key = 'mouse',
target_key = 'hamster',
)
model_hamster.load('context', save_key='_mouse')
model_hamster.train_target(25, save_key='_hamster')
model_hamster.get_representation(eval_model='target', save_libsize=True)
Initializing context scVI model.
Initializing target scVI model.
Loaded /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/config_dict.pkl
Loaded /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_config__mouse.pkl
Loaded /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_optimizer__mouse.opt
Loaded /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_encoder_outer__mouse.pth
Loaded /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_decoder__mouse.pth
Loaded /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_encoder_inner__mouse.pth
Loaded /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/context_lib_encoder__mouse.pth
Training on the target dataset for 25 epochs (= 1150 iterations).
Progress: 99.7% - ETA: 0:00:00 - Epoch: 25 - Iteration: 1146 - ms/Iteration: 33.36 - nELBO: 1752.2 (+2.446) - nlog_likeli: 1264.0 (-1.236) - KL-Div z: 14.263 (-0.047) - KL-Div l: 2.7568 (-0.001) - Align-Term: 471.15 (+3.730). Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/config_dict.pkl
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_config__hamster.pkl
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_optimizer__hamster.opt
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_encoder_inner__hamster.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_encoder_outer__hamster.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_decoder__hamster.pth.
Saved /Users/cschaech/Desktop/scpecies_package/docs/source/tutorials/params/target_lib_encoder__hamster.pth.
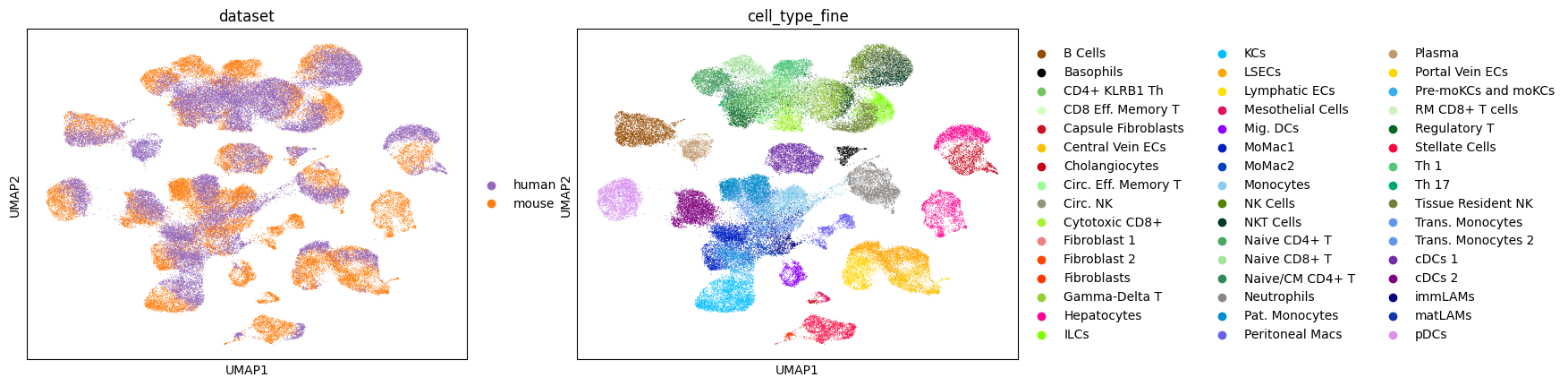
Visualizing the aligned latent space
[18]:
adata_concat = ad.AnnData(
X=sparse.vstack([mdata.mod['mouse'].obsm['z_mu'], mdata.mod['human'].obsm['z_mu'], mdata.mod['hamster'].obsm['z_mu']]).toarray(),
obs=pd.concat([mdata.mod['mouse'].obs[['dataset', 'cell_type_coarse']], mdata.mod['human'].obs[['dataset', 'cell_type_coarse']], mdata.mod['hamster'].obs[['dataset', 'cell_type_coarse']]])
)
# Color scheme for the liver cell dataset. Won't return nice results for other datasets.
palette = return_palette(list(adata_concat.obs.cell_type_coarse.unique()) + list(adata_concat.obs.dataset.unique()))
sc.pp.neighbors(adata_concat)
#neighbors_workaround(adata_concat)
sc.tl.umap(adata_concat)
sc.pl.umap(adata_concat, color=['dataset', 'cell_type_coarse'], palette=palette)